Oxidative wet-chemical graphite delamination is a promising method for large-scale graphene production. Ab initio molecular dynamics simulations show that ideal stacking and oxidation of the graphite layers reduce the friction of sulfuric acid molecules, thereby facilitating intercalation.
Motivation and problem definition
Over the past decade, graphene has attracted a lot of attention in physics, chemistry and materials science due to its outstanding electronic, mechanical and chemical properties. Chemical vapor deposition on a metal substrate is currently the method of choice for preparing large areas of high quality graphene. However, the overall amount of synthesized graphene remains rather small. An alternative process, which has the potential for large-scale graphene production for industrial applications, is the wet-chemical delamination of graphite. A widely used approach is based on the so-called Hummers’ method, which consists of the following steps: first graphite is intercalated by concentrated sulfuric acid. Then, the graphite intercalation compound (GIC) is oxidized. Finally, graphene oxide (GO) layers are separated in solution by hydrolysis and the post-processing of the GO with a reducing agent yields graphene. The overall process, however, is not well understood and is hampered by defect formation. Molecular dynamics (MD) simulations can provide new insights on the atomic scale how oxidation and perturbations in the graphite lattice influence the overall intercalation process.
Methods and codes
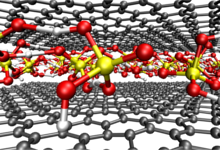 To this end, we performed ab initio molecular dynamics simulations within the Car-Parrinello framework using the CPMD software package. In a recent KONWHIR project Tobias Klöffel, in collaboration with Gerald Mathias from LRZ, significantly improved the OpenMP and MPI parallelization of CPMD. These code modifications clearly boosted the performance, enabling us to treat quite large systems (unit cells with more than 500 atoms) in a reasonable amount of time. For typical simulations on 9 Emmy nodes (180 processes), the improved OpenMP parallelization saved up to 40 % of computer time.
To this end, we performed ab initio molecular dynamics simulations within the Car-Parrinello framework using the CPMD software package. In a recent KONWHIR project Tobias Klöffel, in collaboration with Gerald Mathias from LRZ, significantly improved the OpenMP and MPI parallelization of CPMD. These code modifications clearly boosted the performance, enabling us to treat quite large systems (unit cells with more than 500 atoms) in a reasonable amount of time. For typical simulations on 9 Emmy nodes (180 processes), the improved OpenMP parallelization saved up to 40 % of computer time.
Energies and forces were determined from quantum-chemical density-functional theory calculations employing Vanderbilt ultrasoft pseudopotentials, the PBE exchange-correlation functional and Grimme D2 dispersion corrections. The simulations were done in the canonical ensemble close to room temperature conditions by using Nose-Hoover thermostats. A typical simulation for a trajectory of 70 ps requires 700.000 successive MD steps (each taking about 6 seconds on 9 Emmy nodes), resulting in a total simulation time of roughly 7 weeks. From the MD trajectory we determined equilibrium distributions of atoms (which can be converted into free energy profiles), diffusion coefficients, pair correlation functions or the friction coefficient, which describes the resistance for the sulfuric acid molecules to move between the carbon layers of the graphite crystal. It turned out that especially the computation of a converged friction coefficient requires well-equilibrated systems and quite long simulation times of several ten picoseconds.
Results
Our main results were obtained from simulations of three model setups of the sulfuric acid graphite intercalation compound. Friction coefficients can be extracted from the plateau value of the Green-Kubo force auto-correlation function (see Figure). 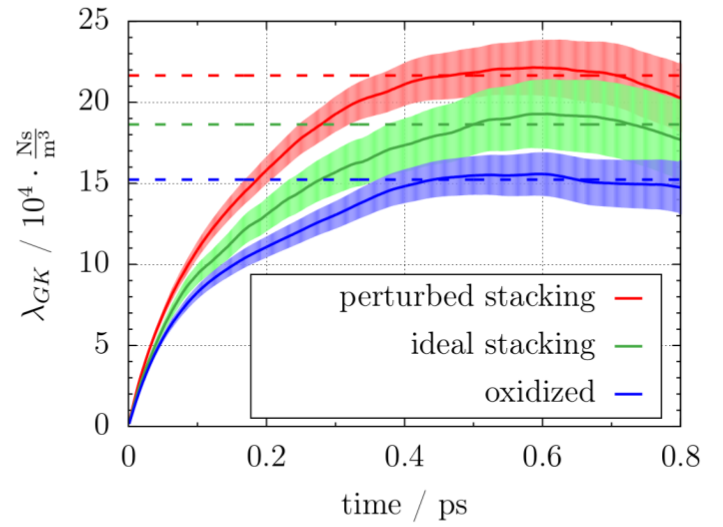 The plot shows that friction is significantly lowered in graphites with ideal AB stacking (see green line) and after oxidation (see blue line) as compared to the setup with perturbed stacking of the graphite layers (see red line). These observations are corroborated by the sulfur atom diffusion coefficient and the lateral distribution of the oxygen atoms in the simulation box and they are explained by a detailed analysis of free energy profiles and the electronic structure. Altogether, we can conclude that the intercalation process strongly benefits from an initial oxidation and a high crystallinity of the graphite lattice. This explains experimental observations on the different behavior of different types of natural graphites.
The plot shows that friction is significantly lowered in graphites with ideal AB stacking (see green line) and after oxidation (see blue line) as compared to the setup with perturbed stacking of the graphite layers (see red line). These observations are corroborated by the sulfur atom diffusion coefficient and the lateral distribution of the oxygen atoms in the simulation box and they are explained by a detailed analysis of free energy profiles and the electronic structure. Altogether, we can conclude that the intercalation process strongly benefits from an initial oxidation and a high crystallinity of the graphite lattice. This explains experimental observations on the different behavior of different types of natural graphites.
Outreach
This work has been published in Nature Communications 9 (2018) 836 (DOI: 10.1038/s41467-018-03211-1) and it will be part of Steffen Seiler’s PhD thesis. The project is financed by the Collaborative Research Center SFB 953 “Synthetic Carbon Allotropes”.
Researcher’s Bio and Affiliation
Steffen Seiler obtained his Master’s degree in Molecular Science at the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) and he is currently a PhD student in the group of Prof. Bernd Meyer at the Interdisciplinary Center for Molecular Materials (ICMM) and the Computer-Chemistry-Center (CCC).
Contact:
Steffen Seiler Interdisciplinary Center for Molecular Materials (ICMM) Friedrich-Alexander-Universität Erlangen-Nürnberg
Mainly used HPC resources at RRZE
Emmy cluster