The histamine H1 receptor (H1R) plays an important role in the context of hypersensitivity reactions and is therefore a main target of antiallergic drugs. Using molecular dynamics (MD) simulations, we intend to unveil details of histamine binding to the H1R and the associated activation of the receptor.
Motivation and problem definition
An excessive stimulation of the H1R by its physiological ligand histamine is coupled to some key symptoms of allergies such as sneezing, pruritus, overproduction of mucus, or even severe reactions like anaphylactic shock. For the design of interfering medications, it is an important prerequisite to gather precise knowledge about the molecular processes involved. Especially the histamine binding pose and the specific underlying interactions with the receptor are of particular interest for pharmaceutical research. Since there exists no crystal structure of the H1R in complex with histamine, we used conventional MD simulations to simulate spontaneous binding of histamine to the receptor and to characterize the binding site in more detail. Moreover, we are currently performing Gaussian accelerated MD (GaMD) simulations to study the conformational changes which might arise upon binding, i.e. the receptor activation which is necessary for downstream intracellular signaling.
Methods and codes
For conventional MD simulations, we mainly use the GROMACS program suite, which offers exceptionally good scaling on MPI-parallel CPU nodes. With 25 nodes on Meggie, an average simulation speed of more than 200 ns/day can be obtained for GPCR systems. Moreover, we are currently running GaMD simulations implemented in AMBER. GaMD is an enhanced sampling algorithm which allows to overcome barriers in the free energy landscape by addition of a Gauss-shaped bias potential to the energy function of the system. Thereby, structural changes that would otherwise be very rare may be observed in significantly reduced simulation time. AMBER can be efficiently run on single GPUs combining thus high simulation speed with less energy consumption and a decreased use of computational resources.
Results
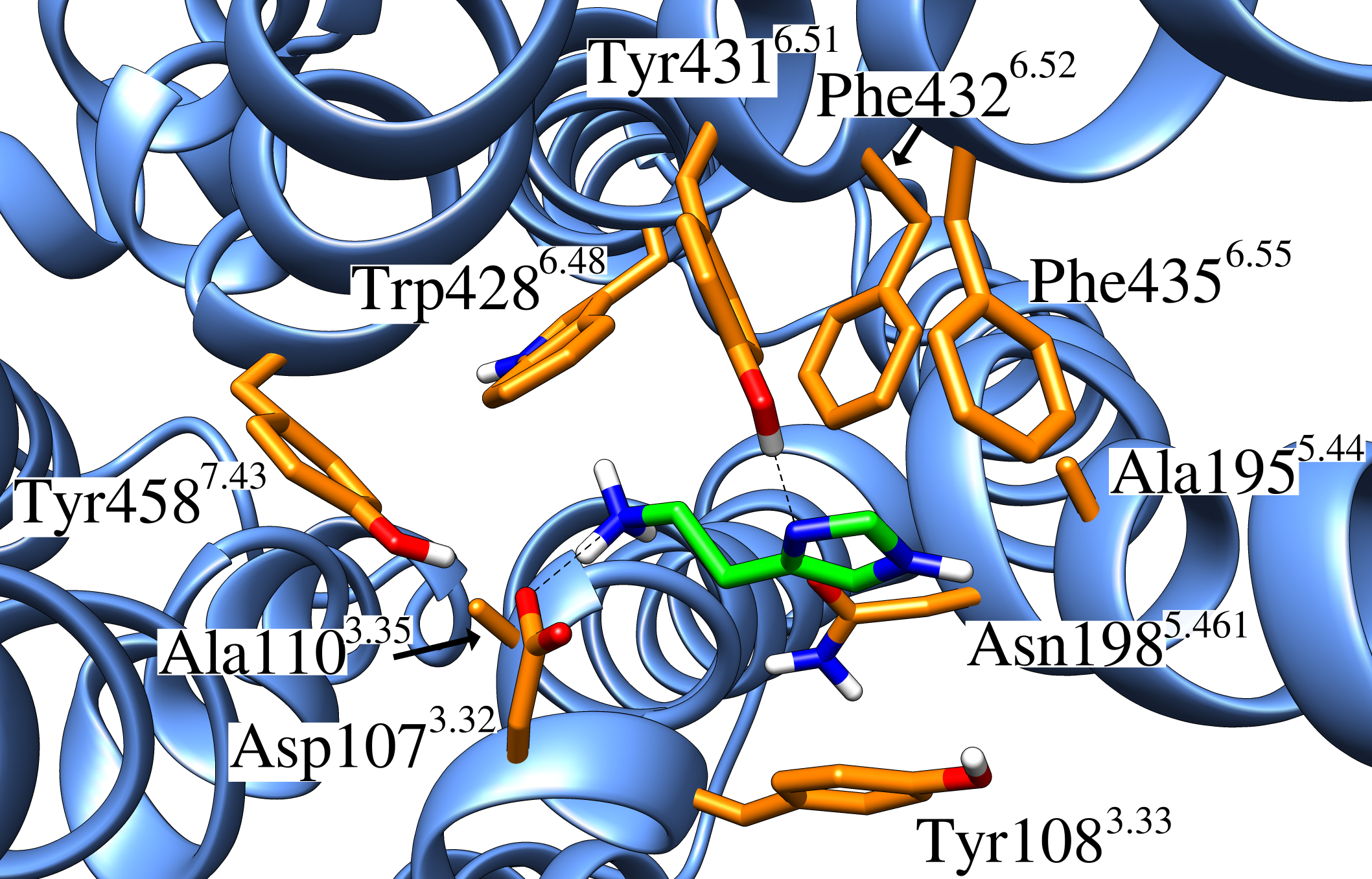
Using conventional MD simulations, we were able to observe a spontaneous binding event of histamine to the H1R. After initial contacts with the extracellular loop 2, the ligand moved first into the vestibule of the binding pocket and then further downwards to the orthosteric binding site. Additional equilibrium MD simulations on a microsecond time scale were used to prove that the determined binding mode is stable and to quantify the most important interacting residues. The obtained data are in good agreement with previous docking studies and site-directed mutagenesis experiments. The final binding mode was shown to be especially characterized by a salt bridge between the ammonium nitrogen atom of histamine and the conserved Asp1073.32. To analyze the relevance of this particular residue for ligand binding, we also performed MD simulations of a histamine-bound D107A mutant receptor, in which we observed that the ligand was much more flexible and that it even dissociated in one of the runs.
Outreach
Christian A. Söldner, Anselm H. C. Horn, Heinrich Sticht, “Binding of histamine to the H1 receptor—a molecular dynamics study”, J Mol Model 2018, 24: 346, DOI: 10.1007/s00894-018-3873-7
Researcher’s Bio and Affiliation
Christian A. Söldner obtained his degree in Molecular Medicine at FAU. After his master’s thesis in the bioinformatics group at the medical faculty’s institute of biochemistry (Prof. Heinrich Sticht), he is currently striving to finalize his Ph.D. thesis in the same group. As a graduate of the GRK1910, his research is mainly focused on the analysis of receptor-ligand interactions.