HPC User Report from F. Weber (Chair of Applied Mechanics)
Investigation of the Influence of Simulation Box Size and Polymer Chain Length on Molecular Dynamics Simulations of Polystyrene
Coarse-grained Molecular Dynamics (MD) simulations were conducted in order to clarify the question if a variation of the number of particles at equal chain lengths has a significant effect on the simulated material behavior, i.e. if the simulation wall time can be reduced. The resulting smaller and therefore more efficient simulation systems were then used to determine the influence of the macro-molecules‘ length when the number of particles is kept equal.
Motivation and problem definition
In [1] uniaxial deformation, relaxation and creep tests on MD systems modeling atactic Polystyrene (PS) have been conducted and discussed. However, the employed simulation box size results in highly time-consuming and data-intensive experiments. Starting from this point, it was tested if a reduction in the number of super-atoms/the simulation box size leads to noticeably different results. The other issue of this project was how severely a reduction in the number of super-atoms per polymer chain affects the material behavior in terms of e.g. a loss of strength and stiffness expected due to reducing structural reinforcements by a lower density of bonds as well as less hooks and entanglements between the molecules.
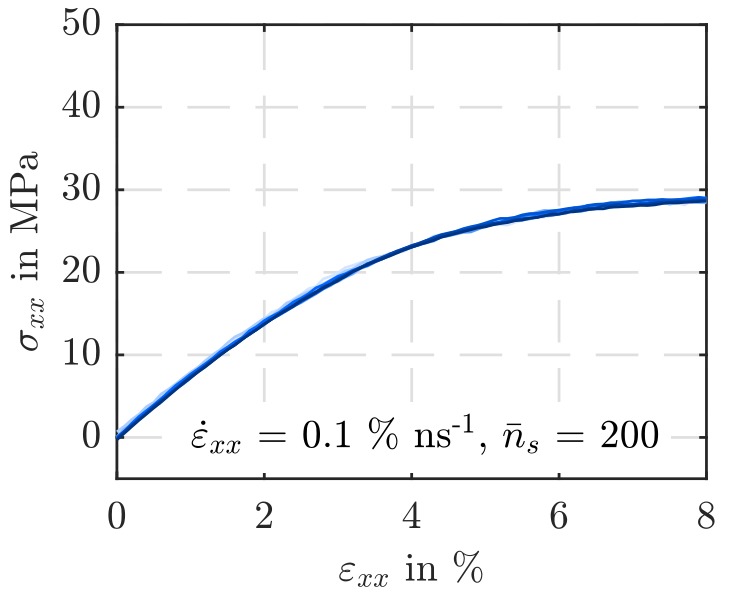
To gain these insights, uniaxial tension simulations were chosen as an exemplary load case, whereby also preceding equilibration simulations of the systems were examined
Methods and codes
The simulation tool IBIsCO[2]developed at the Theoretical Physical Chemistry Group (TPCG) of the Technische Universität Darmstadt (TUDA)together with a governing bash script introduced by the Chair of Applied Mechanics and realizing the computations on the Emmy cluster were employed for the MD simulations.
Results
On the whole, no significant evidence could be detected for a dependence of the averaged simulation output on the particle number. It was possible to reduce the number of super-atoms from 60000 to 10000, whereby both the simulation wall time and the disk usage scale linearly. However, it must be kept in mind that the behavior of smaller simulation boxes is more perceivably determined by local abnormalities. In contrast, the impact of a shortening of the polymer chains on the simulated material’s strength and stiffness is significant.
Outreach
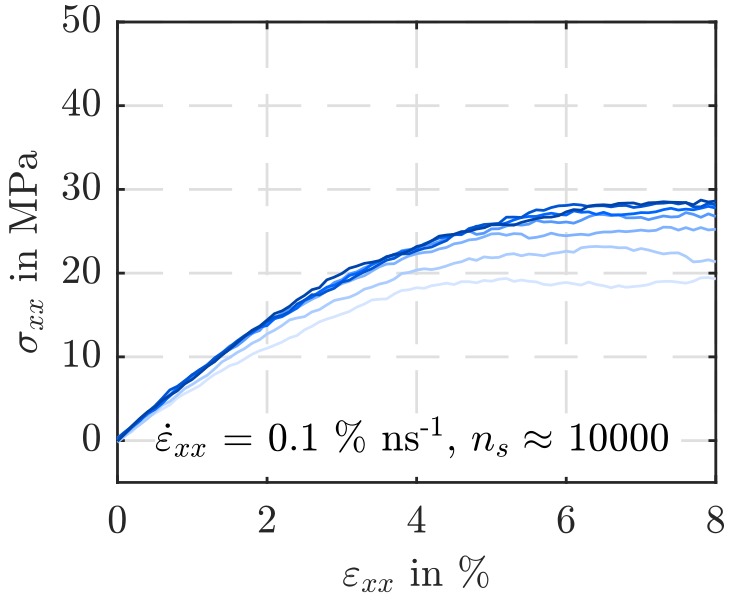
For example, the findings obtained from the variation of the polymer chain length are of interest for the Capriccio method introduced in [3], a partitioned-domain multiscale simulation technique which embeds a region described with MD into a continuum domain reproduced by means of the Finite Element Method (FEM). In this technique a cutting of molecules is necessary to perform the coupling of the two domains.
References
- M. Ries, P. Steinmann, & S. Pfaller. Characterization of the Mechanical Behavior of Polystyrene under Uniaxial Deformation using Molecular Dynamics. Manuscript in preparation. 2019.
- H. A. Karimi-Varzaneh et al. IBIsCO: A molecular dynamics simulation package for coarse-grained simulation. In: Journal of Computational Chemistry 32.7 (2011), pp. 1475-1487.
- S. Pfaller. Multiscale Simulation of Polymers – Coupling of Continuum Mechanics and Particle-
Based Simulation. Doctoral thesis. In: Schriftenreihe Technische Mechanik. Vol. 16. Universität
Erlangen-Nürnberg, 2015.
Researcher’s Bio and Affiliation
Felix Weber is studying Mechanical Engineering at the Friedrich-Alexander University Erlangen-Nürnberg. The presented simulations served his bachelor thesis under the supervision of Maximilian Ries M.Sc. at the Chair of Applied Mechanics, where he is currently working as a research assistant within the context of the Research Training Group GRK 2423 FRASCAL.