The biological activity of G protein-coupled receptors, transmembrane proteins targeted by 36% of drugs, is modulated by their dimerization. Unraveling the dimer interfaces enables us to understand the receptor molecular interactions and thus to modulate their pharmacological effects.
Motivation and problem definition
At first we have studied the heterodimerization of chemokine receptors CXCR4, CCR2, and CCR5 and the effect of cholesterol thereon. Dimers of chemokine receptors play important roles in the human immune systems but they are also involved in HIV infection and in cancer. Moreover, the activity of chemokine receptors is effected by membrane cholesterol. For the development of drugs, specifically targeting chemokine dimers, structural information about their dimerization patterns is required. Unfortunately, experimental data on the possible dimerization interfaces is strongly restricted.
In the second part of the project we have investigated the bivalent ligand binding to the heterodimers formed by the opioid receptors mu (MOR) and delta (DOR). Bivalent ligands consist of two linked ligands targeting two receptors. The common opinion is that the individual ligands are buried in the pockets of the receptors while the linker is located in the extracellular space. The crosslinking of the inactive DOR with the active MOR was shown to significantly reduce tolerance and dependence caused by MOR activators (e.g. morphium). Interestingly, the efficiency of the bivalent ligands depends on the length of the connector. Thereby, the most efficient linkers are too short to bridge the receptors through the extracellular space. Discovery of the binding poses of the bivalent ligand to the heterodimer and the path by which the connector connects the receptors is expected to enable design of pain killers with reduced side effects.
Methods and codes
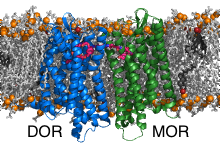
GPCR dimerization is studied by performing multiscaling molecular dynamics simulations using the freeware GROMACS. In the first step, ensembles (consisting typically of ~500 parallel independent runs) of molecular dynamics simulations at coarse-grained resolution are prepared, executed, and analysed by our methodology named DAFT [1]. The most significant dimers are then converted back to atomistic resolution using our program backward [2] and simulated atomistically to refine the interactions at atomistic detail and to enable ligand binding.
Therefore our computational needs are twofold. The ensembles of coarse-grained molecular dynamics simulations require few cores per a single run but a high throughout computing. The atomistic simulations are on the other hand only feasible by using highly parallel computing.
- T.A. Wassenaar, K. Pluhackova, A. Moussatova, D. Sengupta, S.J. Marrink, D.P. Tieleman, R.A. Böckmann. “High-Throughput Simulations of Dimer and Trimer: Assembly of Membrane Proteins. The DAFT Approach.” J. Chem. Theory Comput. 11:2278-2291 (2015)
- T.A. Wassenaar, K. Pluhackova, R.A. Böckmann, S.J. Marrink, D.P. Tieleman. “Going Backward: A Flexible Geometric Approach to Reverse Transformation from Coarse Grained to Atomistic Models.” J. Chem. Theory Comput. 10:676-690 (2014)
Results
Our MD simulations showed similar homodimerization patterns for the closely related CC chemokine receptors in cholesterol-free membranes. Interestingly, cholesterol appeared to specifically effect CC chemokine homodimerization, thus differentiating the close related receptors from each other. Moreover, the heterodimerization patterns with CXCR4 and their cholesterol sensitivity differed. Therefore, apart from generating detailed structural information about chemokine homo and heterodimers, our work sheds first light on the diversifying effects of heterodimerization and the influence of membrane surroundings on closely related receptors.
MOR and DOR form dimers exclusively by interfaces containing helices TM1 or TM5, thereby all possible interface combinations were observed. The different dimers display different linker lengths that would be required to connect the docked ligands. The shortest theoretical linker length is obtained for TM5/TM5 dimers. To these dimers it was possible to model the 19-atom long linker which passes between the helices TM5 and TM6 of the receptors and connects them directly through the lipid bilayer.
Outreach
A publication “Closely related yet unique: Distinct homo- and heterodimerization patterns of G protein coupled chemokine receptors and their fine-tuning by cholesterol” by S. Gahbauer, K. Pluhackova, and R. A. Böckmann, was submitted to PlosCB.
Both projects were presented by S. Gahbauer and K. Pluhackova at 19th IUPAB congress and 11th EBSA congress in Edinburgh in July 2017 and at the GLISTEN Symposium in Porto in March 2017.
The chemokine project is supported by the German Science Foundation (DFG) within the Research Training Group 1962 – Dynamic Interactions at Biological Membranes and the emerging field initiative at the Friedrich-Alexander University Erlangen-Nürnberg (FAU) in Synthetic Biology.
Researcher’s Bio and Affiliation
Kristyna Pluhackova graduated in Chemistry at the Charles University in Prague in 2009. She received her PhD degree in 2015 from FAU Erlangen-Nürnberg for developing methodologies and force fields for membrane-protein simulations in the Computational Biology group of Prof. Rainer A. Böckmann. She is currently a PostDoc in the same group. In the application of molecular dynamics she continues to see the biophysical and biological problems by the eyes of a chemist, interested mainly in the fascinating molecular details. K. Pluhackova has obtained in 2016 a junior investigator grant of the Biology Department at FAU and in fall 2017 she was awarded a research stipend from the German Scientific Foundation to join the experimental group of Prof. Müller at ETH Zürich for two years.
Contact:
Dr. Kristyna Pluhackova Computational Biology Group Friedrich-Alexander-Universität Erlangen-Nürnberg
Mainly used HPC resources at RRZE
large-scale project on Meggie