HPC User Report from A. Leonardi (Institute for Multiscale Simulation)
Engineering Lattice Distortion in Nanomaterials
Understanding the correlation between nanostructure, lattice distortion,and chemical-physical performance is important for the application of nanocrystals. We utilize simulations to design and assist in the synthesis of shape-controlled core@shell nanocrystals and to characterize their internal microstructure.
Motivation and problem definition
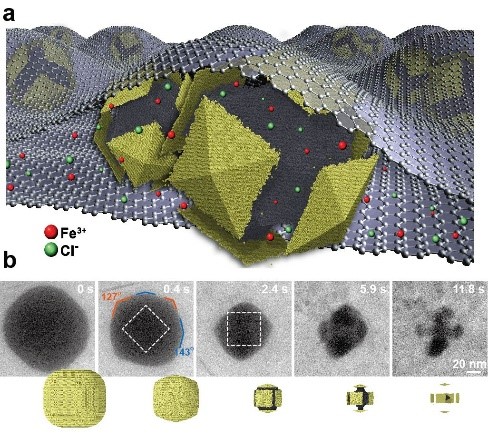
Nanomaterials show chemical-physical properties that are desirable for applications in heterogeneous catalysis, energy storage, plasmonics, and photonics. Distortion of lattice structure is a strategy to taylor these properties and to achieve optimal performance. We address two basic research questions:How can the kinetics of nanocrystal formation be controlled and, therefore, the microstructure of multicomponent materials at the nanoscale be tuned? How does lattice distortion affected microstructure and ultimately the response to external stimuli? These questions are answered with the help of computer simulations. We improve the accuracy and reliability of our simulations by coupling them closely to experiments.
Methods and codes
We use kinetic Monte Carlo (MC) and molecular dynamics (MD) to tackle the multiscale nature of structure distortion and investigate chemical processes that form nanostructured materials. Whereas MC accesses the long-time scale of the kinetics of structure formation, MD provides information on lattice distortion at the atomic scale. We combine self-designed algorithms (Rose-X) and established software applications (LAMMPS) to analyze experimental data and predict the formation of nanostructures.
Results
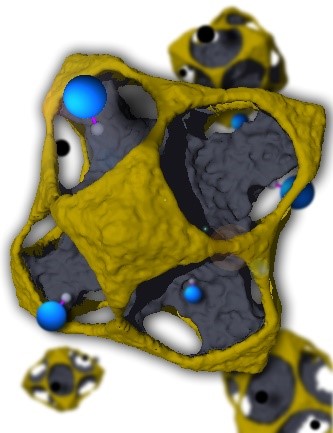
Our MC simulations demonstrate the appearance of characteristic non-equilibrium intermediate microstructures from the etching of core@shell nanocrystals[1]. Convex platelet, concave polyhedron, pod, cage, and strutted-cage shapes are obtained at room temperature with a fully coherent structure exposing crystallographic facets and chemical elements along with distinct particle crystallographic directions. Experiments validated our prediction, although they reveal dissolution rates different from prediction[2]. Dissolution of distinct regions in bi-metallic nanocrystals is affected by partial electrochemical passivation. We couple transmission Electron Microscopy images and MD simulations to elucidate the lattice relaxation as a function of lattice mismatch and shell thickness of core@shell nanocrystals. The enhanced durability of nanocatalyst with intermetallic core[3] and the effect of lattice mismatch and shell thickness on strain [4] was probed with MD simulations
Outreach
-
- A. Leonardi, M. Engel, Particle Shape Control via Etching of Core@Shell Nanocrystals, ACS Nano 12(2018) 91862.
- J.T.L. Gamler, A. Leonardi, H.M. Ashberry, N.N. Daanen, Y. Losovyj, R.R. Unocic, M. Engel, S.E. Skrabalak, Achieving Highly Durable Random Alloy Nanocatalysts through Intermetallic Cores, ACS Nano 13(2019), 4008-40173.
- J.T.L. Gamler, A. Leonardi, X. Sang, K.M. Koczkur, R.R. Unocic, M. Engel, S.E. Skrabalak, Effect of Lattice Mismatch and Shell Thickness on Strain in Core@Shell Nanocrystals, Nanoscale Adv. (2020), https://doi.org/10.1039/D0NA00061B.
- Lei, A. Leonardi, M. Engel, X. Ye, In-Vivo Electron Microscopy of Core-Shell Nanocrystal Etching, preprint (2020)
Researcher’s Bio and Affiliation
Dr. Alberto Leonardi defended his Ph.D. in 2012 at the University of Trento (Italy) with thesis title: “Molecular dynamics simulations and X-ray powder diffraction simulations”. After 2 years of postdoc in continuation of the Ph.D., he moved to Indiana University Bloomington (USA) from 2015 to 2016. In 2017 he joined the research group of Prof. Michael Engel at the Institute for Multiscale Simulation in the Department of Chemical and Biological Engineering at FAU where this work was performed.