HPC User Report from O. Malcioglu (Theoretische Festkörperphysik)
Porphyrins on Surfaces and their Electrophysical Properties
Motivation and problem definition
Porphyrins are intensely colored natural pigments that we owe the green in our world, and the red in our blood to. Wherever and whenever there is life, some form of this “dye of life” is present. This is not surprising, as they are the key ingredients in a wide diversity of important biochemical reactions. Of particular interest is the biological redox processes, such as the conversion of sunlight into chemically stored energy in chlorophyll. It is astonishing that only small variations on the basic structural theme translates into such diverse photophysical properties, while preserving the broad outline of their family.
An important cornerstone for mimicking the efficiency of nature in harvesting and using the energy is to understand the porphyrin interactions. Porphyrins interacting with surfaces are already used successfully in various applications such as surface catalysis, lighting and energy harvesting [1-5]. Research regarding porphyrin have a huge potential to further improve the efficiency of the devices they are used in, by exploiting the effects seen in biological systems. This will not only lead to bio-compatible, environmentally friendly devices, but also the commercial production costs will also be drastically reduced using solution based manufacture techniques.
Methods and codes
We use an ab initio software package, VASP. VASP is highly parallelized code that can also perform mixed GPU/CPU computations. We perform ab initio canonical ensemble molecular dynamics (MD) calculations in order to understand how the porphyrin interacts with the surface. The electronic structure is described within density functional theory, using various flavours such as hybrid functional HSE06. The long-range tail of the interaction potential is simulated with a variety of different correction schemes. In order to attain chemically meaningful simulation times using ab initio principles, we tune a hybrid OpenMP/MPI parallelization scheme in order to minimize the communication between the nodes while attaining a maximum throughput between memory and the cpu. This type of calculation benefits greatly from the presence of GPU accelerator cards. For accessing the quasiparticle levels at a chemical precision comparable with the NIST database, we use GW approximation of Hedin, in particularly we use a variant of sc-GW originally suggested by van Schilfgaarde et al. and implemented in VASP. This is a calculation only possible in a large computation centre, with a computer resembling a shared memory architecture as much as possible. We require typically around 8-16 Gb of memory per cpu, around 100-200 cpus, and naturally a very fast interconnect between the nodes for this kind of calculations
Results
The ab initio molecular dynamics simulations performed in RRZE revealed a pathway where:
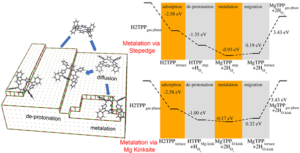
H2TPP self metallates to MgTPP, shedding light to the experiment carried out by our collaborators [6]. In the proposed pathway, the H2TPP gets physisorbed to pristine MgO (100) terrace, migrates to step edges of (100) planes or Mg kink defects due to pedesis, metallates at the defect site, and moves back to MgO (100) terrace as MgTPP. Van der Waals dispersion (DFT-D3) corrections were used to address the free energy contribution. The dynamic hindrance due to phenyl rings explain experimentally observed mobility on the pristine surface [7, 8]. The common MgO (100) defects (the stepedge and the kink) provide the porphyrin with reactive anchor points. When porphyrin encounters such a defect, it rapidly deprotonates, similar to what is observed in the TiO2 surface [9]. The metalation to MgTPP on these sites is energy driven, and once metallated, the barrier of MgTPP migrating back to the pristine (100) terrace is low.
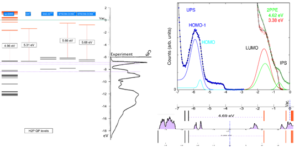
scGW method is a many body perturbative method which is capable of producing chemically accurate (calibrated against NIST) quasiparticle levels. Our comparative analysis of the UPS experiment performed by the Funcos group [10] involved various isomers, conformers, and oligomers of H2TPP and MgTPP. We have found a possible supramolecular network which aligns perfectly with the features seen in the experiment. This supramolecular network exhibits various interesting effects that can easily be used in various commercial applications, most significantly dispersive bands. Such dispersive bands explain increased electroluminescence efficiency recently reported [11].
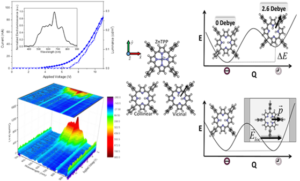
An all-organic, white light emitting device is an important goal in industry both due to economic and environmental reasons. Although H2TPP shows superior electrochemical and photophysical features compared to ZnTPP which has a Zinc atom core, ZnTPP performs better as a typical electroluminescent device. Our collaborators Ruben et al. found out that all organic H2TPP embedded in a matrix feature an unexpected yellowish white EL [12]. Since no degradation upon both charge injection and transport and/or difference in the device mechanism were noted, they postulated a change in the nature of the emitting excited state in H2TPP devices under electrical stimuli. We identified a tautomerization effect that explains the device data they measure. External electric fields have a profound impact on H2TPP while ZnTPP features no change as evidenced by device polarizability. We explain this by the induced local fields, creating a vicinal H2TPP tautomer, where hydrogens of the macrocycle align to counteract the induced local field.
Outreach
The results of these works are presented as oral contributions in APS march meeting 2017, DPG 2016, ECOSS 32 and as posters in PSI-K 2017, and in various departments. Furthermore, one of the publications was selected as the frontispiece of Advanced Functional Materials.
This project is part of the DFG-funded research group FOR 1878 (BO1851/4-1 till March 2017 and FWF I3385-N34 since June 2017 by FWF Austria, via D.A.CH).
Publications that resulted so far from this project:

- Classen, A., et al., Electronic structure of tetraphenylporphyrin layers on Ag(100). Physical Review B, 2017. 95(11).
- Weber, M.D., et al., From White to Red: Electric-Field Dependent Chromaticity of Light-Emitting Electrochemical Cells based on Archetypal Porphyrins. Advanced Functional Materials, 2016. 26(37): p. 6737-6750.
- In preparation: O. B. Malcioglu, Ph. Auburger, M. Bockstedte, Energy driven self-metallation of H2TPP on MgO surface mediated by extended defects.
References
- Friend, R.H., et al., Electroluminescence in conjugated polymers. Nature, 1999. 397(6715): p. 121-128.
- Gottfried, J.M., Surface chemistry of porphyrins and phthalocyanines. Surface Science Reports, 2015. 70(3): p. 259-379.
- Kadish, K.M., K.M. Smith, and R. Guilard, Handbook of Porphyrin Science. 2010 World Scientific.
- Rajapakse, G.V.N., Photophysical properties of metallotetraphenyltetrabenzoporphyrins: Insights from experimental and theoretical studies. 2008, Bowling Green State University.
- Urbani, M., et al., Meso-substituted porphyrins for dye-sensitized solar cells. Chem Rev, 2014. 114(24): p. 12330-96.
- Schneider, J., et al., Porphyrin Metalation at MgO Surfaces: A Spectroscopic and Quantum Mechanical Study on Complementary Model Systems. Chemistry, 2016. 22(5): p. 1744-9.
- Auwarter, W., et al., Porphyrins at interfaces. Nat Chem, 2015. 7(2): p. 105-20.
- Duncan, D.A., et al., Immobilised molecular catalysts and the role of the supporting metal substrate. Chem Commun (Camb), 2015. 51(46): p. 9483-6.
- Lovat, G., et al., Hydrogen capture by porphyrins at the TiO2(110) surface. Phys Chem Chem Phys, 2015. 17(44): p. 30119-24.
- Classen, A., et al., Electronic structure of tetraphenylporphyrin layers on Ag(100). Physical Review B, 2017. 95(11).
- Coutsolelos, A.G., et al., Peripheral substitution of tetraphenyl porphyrins: Fine-tuning of the self-assembling towards enhanced electroluminescence. ChemPlusChem, 2017.
- Weber, M.D., et al., From White to Red: Electric-Field Dependent Chromaticity of Light-Emitting Electrochemical Cells based on Archetypal Porphyrins. Advanced Functional Materials, 2016. 26(37): p. 6737-6750.
Researcher’s Bio and Affiliation