HPC User Report from J. Kaindl (Chair of Medicinal Chemistry)
Allosteric modulation at G protein-coupled receptors
The activation process of G protein-coupled receptors (GPCRs) is not fully understood up to date. Allosteric modulators targeting GPCRs not only represent a group of novel drugs with the potential for increased drug safety but offer the chance to further unravel the process of receptor activation.
Motivation and problem definition
We studied the binding-mode of a negative allosteric modulator targeting the β2 adrenergic receptor (ß2AR) as revealed by X-ray crystallography. The allosteric modulator was discovered by virtual screening in combination with SAR studies. Compared to conventional ligands targeting G protein-coupled receptors (36% of marketed drugs), allosteric modulators bind outside of the highly conserved orthosteric binding pocket. Therefore they have an increased potential for sub-type selectivity. Additionally, allosteric modulators mediate their effect by modulating the response to native hormones and neurotransmitters which can result in an improved drug tolerance. Besides the latter potential therapeutic advantages, allosteric modulators can also be useful tools to study the process of receptor activation. The X-ray structure of the ß2AR revealed a binding-site of the allosteric modulator at the membrane facing surface of the receptor in contact to a second antiparallel symmetry mate (Fig. 1). We wanted to use Molecular Dynamics (MD) simulations to investigate the stability of the proposed binding-mode without the additional crystal contacts.
In other inactive state crystal structures of ß2AR a water molecule is located at this binding-site connecting E122 in transmembrane helix (TM) 3 and V206 and S207 in TM5. In the active state the water molecule is replaced and E122 directly interacts with TM5. As the modulator replaces this water molecule and stabilizes the interaction between TM3 and TM5 around a previously identified molecular switch for receptor activation formed by I121, P211 and F282 the X-ray crystal structure shows how the allosteric modulator can stabilize the inactive state of the receptor. Inspired by this pocket, mutagenesis studies were performed revealing a strong allosteric effect of this binding-site. The Arginine mutant was insensitive to agonist activation while the Glutamine mutant was more sensitive to receptor activation. To gain insight on the nature of the allosteric modulation on the molecular level we performed additional MD simulations of wild-type ß2AR (E122) and its mutants E112Q and E122R in the presence of the connecting water molecule.
Methods and codes
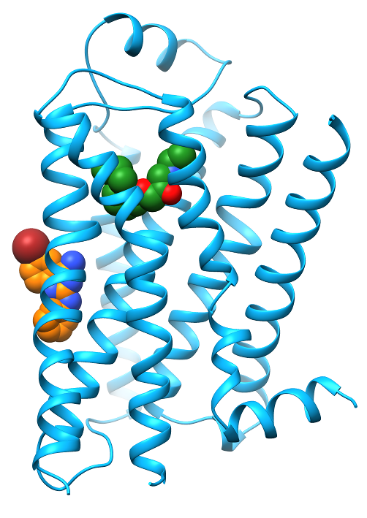
To test the stability of the binding-mode of the allosteric modulator revealed by X-ray crystallography we performed 12µs (3 x 4µs) atomistic MD simulations. The influence of the introduced mutations on interaction between TM3 and TM5 around the activation switch were analyzed with additional 4µs (2 x 2µs) MD Simulations, each. We applying the free open-source software GROMACS utilizing the general amber force filed (GAFF), the lipid14 force field and the ff14SB force field for small molecules, lipids and protein, respectively. The simulation systems contained about 70000 atoms. MD simulations of this scale are only feasible when utilizing highly parallel computing.
Results
The simulations of the described ligand receptor complex revealed a stable binding mode at the membrane facing surface of the receptor. The simulations confirmed that the ligand also binds to the receptor without a second protein stabilizing the binding mode like it is in the crystal structure. The simulations of the wild-type ß2AR (E122) revealed a stable water mediated interaction of E122 of TM3 and V206 and S207 of TM5 as proposed by X-ray crystallography. While the Q122 forms less stable interactions to TM5 suggesting a higher sensitivity to receptor activation, R122 directly interacts with TM5 which may prevent the rearrangement necessary for receptor activation and therefore stabilizing the inactive state. The combination of Virtual screening, SAR studies, X-ray crystallography and MD simulations gave new insights in the activation process of the ß2AR.
Outreach
A publication “Discovery of an allosteric modulator binding to a conformational hub” is in preparation.
The project was supported by the German Science Foundation (DFG) within the Research Training Group 1910 – Medicinal Chemistry of Selective GPRC Ligands.
Researcher’s Bio and Affiliation
Jonas Kaindl graduated in Pharmacy at the FAU in 2014. Since then he does his PhD in the Medicinal Chemistry group of Prof. P. Gmeiner. Jonas Kaindl is a member of the Research Training – Medicinal Chemistry of Selective GPCR Ligands (GRK1910) funded by the German Research Foundation (DFG).