HPC User Report from T. Klöffel (Interdisciplinary Center for Molecular Materials)
Deactivation of Hazardous Chemicals at Oxide Surfaces
The catalytic deactivation of hazardous chemicals at oxide surfaces is investigated using ab initio Car-Parrinello molecular dynamics simulations in order to explore the largely unknown chemistry of these compounds.
Motivation and problem definition
After World War II, large quantities of highly toxic chemical warfare agents, such as sulfur mustard, were disposed in the Baltic Sea where they now constitute a major environmental hazard. New fast, secure, and non-destructive methods are required for the decontamination of highly corroded containers, which are nowadays frequently found in fishing nets or at beaches of the Baltic sea. A very promising approach is to use oxides as catalysts for the decomposition of the chemical warfare agents via hydrolysis reactions. In particular ZnO nanorods are promising candidates according to recent experimental work. However, a detailed understanding of the chemical process of defunctionalization at the solid/liquid interface is mostly lacking, as it is close to impossible to perform standard surface science experiments due to the high toxicity of the materials. Yet, basic insights into the reaction mechanisms at the surface are crucial for improving the catalyst performance. For such hazardous materials in silico research can greatly reduce the number of required experiments.
Methods and codes
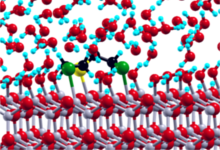 In the first stage of the project high-throughput static DFT calculations employing the PWscf code (www.quantum-espresso.org) were performed to screen adsorption geometries of sulfur mustard molecules on ZnO surfaces in vacuum. In the second stage, the dynamics and first elementary decomposition steps of the sulfur mustard molecule in liquid water were studied using Car-Parrinello molecular dynamics (CPMD). Finally, the combined system consisting of a ZnO catalyst surface, a sulfur mustard molecule and an explicit water environment is considered. Ab initio CPMD simulations for such large systems (more than 600 atoms and 3300 electrons) became only possible after extensive improvement of the OpenMP and MPI parallelization of the CPMD code, which was done in close collaboration with Gerald Mathias from LRZ München in the framework of a KONWHIR project.
In the first stage of the project high-throughput static DFT calculations employing the PWscf code (www.quantum-espresso.org) were performed to screen adsorption geometries of sulfur mustard molecules on ZnO surfaces in vacuum. In the second stage, the dynamics and first elementary decomposition steps of the sulfur mustard molecule in liquid water were studied using Car-Parrinello molecular dynamics (CPMD). Finally, the combined system consisting of a ZnO catalyst surface, a sulfur mustard molecule and an explicit water environment is considered. Ab initio CPMD simulations for such large systems (more than 600 atoms and 3300 electrons) became only possible after extensive improvement of the OpenMP and MPI parallelization of the CPMD code, which was done in close collaboration with Gerald Mathias from LRZ München in the framework of a KONWHIR project.
Results
The static DFT calculations provided a basic understanding of the molecule–substrate interactions. By using accelerated molecular dynamics techniques (well-tempered metadynamics), first decomposition steps of sulfur mustard molecules in water could be observed. Calculations of the reaction free energy barriers are in progress. The adsorption of sulfur mustard from liquid water onto the ZnO surfaces is currently investigated using umbrella sampling. Well-tempered metadynamics simulations already provided first insights into the decomposition of pre-adsorbed sulfur mustard molecules on the ZnO surface.
Outreach
This work is supported by the Cluster of Excellence EXC 315 “Engineering of Advanced Materials” (EAM). Additional computer time was provided by a “Summer of Simulation 2017” project of LRZ München. Results of the simulations were presented at the WATOC 2017 conference in München and the annual Spring Meeting of the Deutsche Physikalische Gesellschaft in Berlin. Two publications and a contribution for the Supercomputing Conference SC18 in Dallas, Texas (together with Gerald Mathias from LRZ) are currently in preparation. The simulations will form a main part of Tobias Klöffel’s PhD thesis.
Researcher’s Bio and Affiliation
Tobias Klöffel is a PhD student in the group of Prof. Bernd Meyer at the Interdisciplinary Center for Molecular Materials (ICMM) and the Computer-Chemistry-Center (CCC). He has studied Chemistry at the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) and received his Master’s degree in 2013.