HPC User Report from P. Schwarz (Interdisciplinary Center for Molecular Materials)
Chemical Functionalization of Oxide Surfaces
The chemical reaction of functional linker molecules with aluminum oxide surfaces in a liquid environment at finite temperatures is studied by ab initio molecular dynamics simulations. The simulations provide a detailed understanding of reaction mechanisms at the solid/liquid interface in which the solvent molecules are actively involved.
Motivation and problem definition
The functionalization of oxide surfaces by molecules plays an important role in many areas, such as nanoparticle synthesis, molecular electronics or in the fabrication of hybrid organic/inorganic solar cells. The attached molecules can protect the underlying surface from chemical attack, they can alter material’s properties, e.g. the work function or electron injection barrier, or they can act as functional units themselves, e.g. as conduction channel in a molecular field-effect transistor. Surface functionalization is mostly done by wet-chemical processes in which molecules are attached by condensation reactions, i.e by elimination of water molecules. While adsorption of molecules from the gas phase has been studied extensively, mechanisms of chemical reactions at the solid/liquid interface are basically unexplored. Our simulations therefore aim at a first fundamental understanding of the important elementary reaction steps when a molecule from solution binds to a solid surface. In particular, we address the role of the solvent and the impact of the surface structure and composition. For our simulations we have chosen aluminum oxide as substrate, isopropanol as the liquid phase and methylsilanetriol as reactive molecule. This is a typical combination often utilized in experiment.
Methods and codes
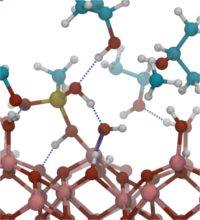 For an unbiased description of the chemical processes we use ab initio molecular dynamics simulations, specifically the Car-Parrinello molecular dynamics (CPMD) method and code. CPMD is based on density functional theory (DFT) as quantum-chemical electronic structure method. Wavefunctions are expanded in a plane-wave basis set and ionic cores are replaced by Vanderbilt ultrasoft pseudopotentials. Van-der-Waals interactions are included by Grimme’s D2 correction scheme. A canonical NVT ensemble is established by Nose-Hoover thermostats and the equations of motion for the atoms and the wavefunctions are integrated with the Verlet algorithm. In order to be able to observe chemical reactions (“rare events”), we apply various accelerated sampling techniques, such as metadynamics, thermodynamic integration and umbrella sampling, which also give information on the free energy surface. Typical simulations (about 250 atoms per unit cell) require about 2.1 s for one MD step on 10 Emmy nodes, i.e. a 100 ps trajectory is obtained in about 4 weeks.
For an unbiased description of the chemical processes we use ab initio molecular dynamics simulations, specifically the Car-Parrinello molecular dynamics (CPMD) method and code. CPMD is based on density functional theory (DFT) as quantum-chemical electronic structure method. Wavefunctions are expanded in a plane-wave basis set and ionic cores are replaced by Vanderbilt ultrasoft pseudopotentials. Van-der-Waals interactions are included by Grimme’s D2 correction scheme. A canonical NVT ensemble is established by Nose-Hoover thermostats and the equations of motion for the atoms and the wavefunctions are integrated with the Verlet algorithm. In order to be able to observe chemical reactions (“rare events”), we apply various accelerated sampling techniques, such as metadynamics, thermodynamic integration and umbrella sampling, which also give information on the free energy surface. Typical simulations (about 250 atoms per unit cell) require about 2.1 s for one MD step on 10 Emmy nodes, i.e. a 100 ps trajectory is obtained in about 4 weeks.
Results
The chemical binding of methylsilanetriol to the alumina surfaces can be divided into two steps: the coordination of the molecule to the surface and the final condensation reaction. While initial surface coordination is spontaneous in vacuum, it becomes hindered in the isopropanol liquid or in the presence of residual water on the alumina surface. For the condensation reaction we could identify two different mechanisms which differ in the relevant transition state: the reaction either proceeds via formation of a five-fold coordinated Si atom or via formation of an intermediate six-membered ring, involving the Si atom, a surface Al atom and the O atom of the leaving water molecule. The condensation reaction is found to be spontaneous under water-free conditions, but becomes activated in the presence of residual water.
Outreach
This work is supported by the Cluster of Excellence EXC 315 “Engineering of Advanced Materials” (EAM). The results were presented at the annual Spring Meeting of the Deutsche Physikalische Gesellschaft and at several EAM symposia. The PhD thesis of Paul Schwarz will be mainly based on the results of these simulations.
Researcher’s Bio and Affiliation
Paul Schwarz received his Master’s degree in Chemistry at the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) in 2013. He is currently a PhD student in Prof. Bernd Meyer’s group at the Interdisciplinary Center for Molecular Materials (ICMM) and the Computer-Chemistry-Center (CCC).