Molecular Dynamics (MD) simulations using new simplified pair-specific ab initio-based molecular models were performed to improve the predictive performance for calculating the dynamical properties of fluid systems over a broad range of system densities.
Motivation and problem definition
For calculating the dynamical properties of fluid systems via MD simulations, accurate molecular models are required. Though such molecular models are based on a physical theory and can often be transferred within certain fluid classes, their optimization requires usually experimental vapor-liquid equilibrium data. An alternative are theoretical simplified pair-specific molecular models developed based on ab initio calculations in the limit of zero-density. However, to which extent such simplified molecular models provide an improvement in predictions over a broad range of system densities is not thoroughly addressed in the literature until now.
Methods and codes
In a collaborative research project funded by the German research foundation (DFG), two research groups headed by Dr. Robert Hellmann (project number DFG/HE 6155/2-1) from the University of Rostock and by Prof. Dr.-Ing. habil. Andreas Paul Fröba (project number DFG/FR 1709/12-1) from the Institute of Advanced Optical Technologies – Thermophysical Properties (AOT-TP) at the Friedrich-Alexander-University Erlangen-Nürnberg (FAU) are involved. The new pair-specific ab initio-based molecular models were obtained by Dr. Hellmann for selected fluid systems and applied for predictive MD simulations performed in Erlangen.
For the latter simulations, the GROMACS 5.1.2 software [1] was used to carry out parallelized computations typically on 3 to 5 nodes on the EMMY cluster. In-house post processing FORTRAN scripts using Open Multi Processing (OpenMP) and Message Passing Interface (MPI) methods were employed for calculating the dynamical properties.
Results
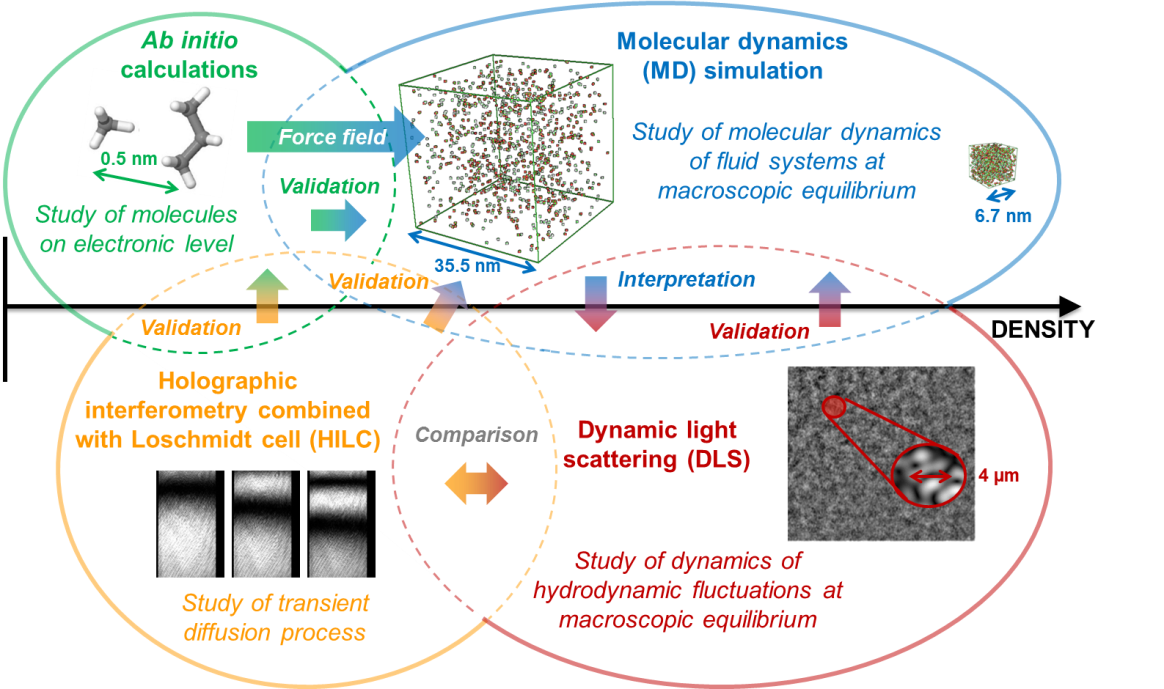
The predictive power of the new ab initio-based molecular models in determining mass diffusivities and viscosity were studied over a broad density range from the superheated vapor over the gas state and supercritical region up to the compressed liquid state. The corresponding results are summarized in Refs. [2-4]. As model systems methane, propane, and carbon dioxide as well as their binary mixtures were investigated with respect to self-diffusivity and viscosity of the pure systems as well as Fick diffusivity of the binary mixtures. A comparison of the dynamical properties for the studied fluid systems obtained from MD simulations showed that the approach of using new ab initio-based molecular models improves the prediction results in most cases compared to the use of commonly employed effective molecular models. This could be demonstrated by comparison with the theoretical ab initio-derived calculations as well as with available experimental data from literature including Fick diffusivities obtained at AOT-TP using optical methods (see Fig. 1).
Outreach
The main scientific findings are published in the form of three journal articles [2-4] in Fluid Phase Equilibria. Furthermore, the results of this project were presented in the form of three oral presentations at the “Thermodynamik-Kolloquium” in Dresden, September 2017, the “Twentieth Symposium on Thermophysical Properties,” Boulder, June 2018, and the “International Conference on Advanced Optical Technologies,” Erlangen, March 2019. Three abstracts in the form of poster contributions were presented or accepted for the “21st European Conference on Thermophysical Properties” in Graz, September 2017, the “Diffusion Fundamentals VIII” in Erlangen, September 2019, and the “Thermodynamik-Kolloquium” in Duisburg, October 2019.
References
[1] M.J. Abraham, T. Murtola, R. Schulz, S. Páll, J.C. Smith, B. Hess, E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX 1-2, 19-25 (2015).
[2] U.A. Higgoda, R. Hellmann, T.M. Koller, A.P. Fröba, Self-diffusion coefficient and viscosity of methane and carbon dioxide via molecular dynamics simulations based on new ab initio-derived force fields, Fluid Phase Equilib. 481, 15-27 (2019).
[3] U.A. Higgoda, R. Hellmann, T.M. Koller, A.P. Fröba, Enhancement of the predictive power of molecular dynamics simulations for the determination of self-diffusion coefficient and viscosity demonstrated for propane, Fluid Phase Equilib. 496, 69-79 (2019).
[4] U.A. Higgoda, C.J. Kankanamge, R. Hellmann, T.M. Koller, A.P. Fröba, Fick diffusion coefficients of binary fluid mixtures consisting of methane, carbon dioxide, and propane via molecular dynamics simulations based on simplified pair-specific ab initio-derived force fields, Fluid Phase Equilib. 500, 112257 (2019).
Researcher’s Bio and Affiliation
Ubaya Higgoda obtained his Master of Science (M.Sc.) degree in Information Technology at the KTH Royal Institute of Technology, Sweden. Since February 2016, he is working as a doctoral candidate at AOT-TP under the supervision of Prof. Dr.-Ing. habil. Andreas Paul Fröba.