Molecular Dynamics (MD) simulations give insight into the microscopic structure, and can be used to calculate various thermophysical properties. In the present work, an evaluation of available force fields (FFs) is performed and the all-atom L-OPLS FF is chosen for further improvement at elevated process-relevant temperatures.
Motivation and problem definition
Systems based on long-chained linear and branched alkanes and alcohols as well as liquid organic hydrogen carriers (LOHCs) are of interest in many areas of chemical and energy engineering. Examples include the synthesis of high-value fuels or usage as novel energy carriers. For such long-chained and bulky compounds, literature data is lacking and existing models fail to properly capture the fluid behavior over the range of process-relevant temperatures and pressures. Information on the thermophysical properties of these substances is imperative for safe and economical process design. In addition to expanding available data through experiments, MD simulations are used to aid in developing new prediction models and to efficiently obtain thermophysical properties for substances or thermodynamic conditions, which are difficult to realize with experiments. The current work is embedded into an ongoing Sino-German joint research project between the collaborating research groups of the Institute of Advanced Optical Technologies – Thermophysical Properties (AOT-TP), at the Friedrich-Alexander University Erlangen-Nürnberg (FAU) headed by Prof. Dr.-Ing. habil. Andreas Paul Fröba, and the Jiaotong University of Xi’an, China, headed by Prof. Jiangtao Wu. The project is funded by the German Research Foundation (DFG), project number DFG FR 1709/15-1, and titled “Thermophysical Properties of Long-Chained Hydrocarbons, Alcohols, and their Mixtures with Dissolved Gases”. The aim is to characterize thermophysical properties of the aforementioned substances reliably and efficiently by creating synergy between experiments and MD simulations.
Methods and codes
All MD simulations in this work are performed with the GROMACS 5.1.2 [1] simulation software, in thermodynamic equilibrium, with about 30,000 interaction sites. While all properties can directly be calculated using GROMACS, the large amounts of data required for the analysis of molecular motion and statistical fluctuations are more efficiently processed with the FORTRAN scripts developed in-group. In this way, the high-performance parallel programming capabilities of the HPC cluster “Emmy”, where all work is performed, can be properly utilized. Typical production jobs parallelize tasks on 5-6 nodes.
Results
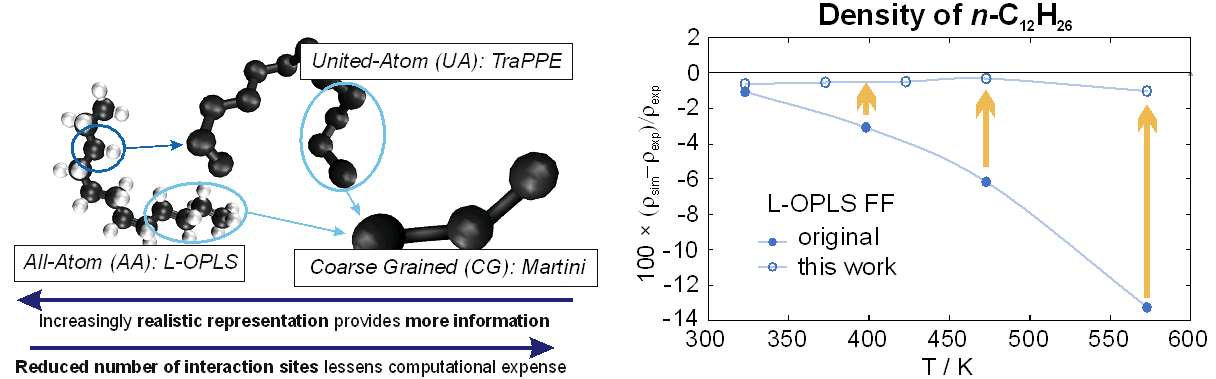
In an effort to improve performance, a force field (FF) evaluation was performed to assess the trade-off between realistic molecular representation and simulation performance for three common FFs from the literature: L-OPLS – all-atom (AA) [2], TraPPE – united-atom [3], and MARTINI – coarse-grained [4], see Figure 1. In order to address the impact of alkane chain length, branching, and hydroxylation, four representative substances were chosen and the viscosity, surface tension, density, and self-diffusion coefficient were obtained for each FF and compared to experimental data. The L-OPLS FF was found to provide the most accurate results for both the equilibrium and dynamic properties for all four representatives and was thus extended to higher temperatures. This improved L-OPLS FF will be used for the remaining substances in this work. The resources provided by the “Emmy” HPC cluster are vital for fast and accurate FF evaluation, the acquisition of reliable data, and insight into the fluid structure.
Outreach
Preliminary results have been published in the International Journal of Thermophysics [5]. Two publications addressing the properties of the pure components are in preparation.
At least three further papers are planned for the three-year project. Furthermore, results of this project were presented in the form of a presentation at the 19th meeting of the International Association for Transport Properties (IATP) in Erlangen, August 2019, and a poster at the international conference Diffusion Fundamentals VIII in Erlangen, September 2019.
References
- Berendsen, H. J. C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43-56
- Siu, S. W. I.; Pluhackova, K.; Böckmann, R. A. Optimization of the OPLS-AA Force Field for Long Hydrocarbons. J. Chem. Theory Comp. 2012, 8, 1459-1479
- Martin, M. G.; Siepmann, J. I. Transferable Potentials for Phase Equilibria. 1. United-Atom Description of n-Alkanes. J. Phys. Chem. B 1998, 108, 750-760
- Marrink, S. J.; de Vries, A. H.; Mark, A. E. Coarse Grained Model for Semiquantitative Lipid Simulations. J. Phys. Chem. B 2004, 108, 750-760
- Koller, T. M.; Yan, S.; Steininger, C.; Klein, T.; Fröba, A. P. Interfacial Tension and Liquid Viscosity of Binary Mixtures of n-Hexane, n-Decane, or 1-Hexanol with Carbon Dioxide by Molecular Dynamics Simulations and Surface Light Scattering. Int. J. Thermophys. 2019, 40, 79
Researcher’s Bio and Affiliation
Frances Lenahan obtained her Master of Science (M.Sc.) degree in Advanced Optical Technologies at the Friedrich-Alexander-University Erlangen-Nürnberg (FAU). Since January 2019, she has been working as a doctoral candidate in the Institute of Advanced Optical Technologies – Thermophysical Properties (AOT-TP) under the supervision of Prof. Dr.-Ing. habil. Andreas P. Fröba.