HPC User Report from F. Beierlein (NHR@FAU, Computer Chemistry Center)
DNA Repair by Thymine DNA Glycosylase (TDG)
Thymine DNA glycosylase (TDG) is an important enzyme involved in DNA repair, which removes mispaired or modified DNA bases and thus ensures genetic integrity. In particular, TDG removes thymine, uracil, and oxidized forms of 5-methylcytosine (5-carboxylcytosine and 5-formylcytosine), which can be formed by a series of methylation and oxidation steps from cytosine.
Motivation and problem definition
TDG removes the mispaired bases by cleaving the glycosidic bond between the sugar and the base of the nucleosides, and the correct base pair is re-established in a process called base excision repair (BER). Interestingly, 5-methylcytosine (mC) itself and its first oxidation product 5-hydroxymethylcytosine (hmC) are not processed by TDG, in contrast to the higher oxidized forms 5-formylcytosine (fC) and 5-carboxylcytosine (caC).
The discrimination between these different forms of (oxidized) methylcytosine (and thus the reason for substrate specificity) can, in principle, occur at 3 different stages: during complex formation between TDG and DNA with the base of interest in its intrahelical form (“flipped-in”), after the base has been flipped out into the enzyme’s active site (extrahelical complex), or at the chemical step (C-N bond dissociation reaction, not covered in this part of the project).
Methods and codes
To study the substrate specificity of TDG we performed extensive atomistic molecular dynamics (MD) simulations with Amber 20[1,2] in which we studied extra- and intrahelical complexes of TDG with DNA (approx. 67,000 atoms) containing caC, fC, hmC and mC, both in their amino- and imino-tautomeric forms, and in different protonation states of the enzyme active site. These MD simulations provide a deep insight into the conformation of the enzyme and the DNA and allow a detailed analysis of the interactions between the extrahelical base and the protein residues in the binding pocket. In addition to conventional, unbiased MD simulations, we employed a rigorous free energy technique called thermodynamic integration (TI), which provides relative binding free energies of the different substrates bonded to TDG.[3] Thermodynamic integration is an example of so-called alchemistic perturbation methods, in which a solute (in our case a DNA base) is “perturbed”, i.e. mutated in silico into another compound, and the associated difference in binding free energy is calculated. To ensure a smooth transition of the initial compound into the other, the perturbation is splitted into a series of nonphysical intermediates, the so-called λ windows. Each of these λ windows represents an individual MD simulation; therefore, all λ windows can in principle be run in parallel, using one GPU for each λ window (or by using loops over subsets of λ windows that run serially/consecutively on one GPU). Using the dual-topology softcore TI implementation in Amber, it is possible to perform perturbations that change the chemical nature of the solute of interest in a much larger degree compared to other (single topology) approaches, an advantage we have exploited in our present research.
Normal, unbiased MD simulations perform very well on GPU systems like the RRZE’s RTX 3080 or Tesla A40 nodes (with a GPU utilization of > 90 % and a gain in simulation time of roughly one order of magnitude compared to a typical MPI CPU-based setup). In addition to the GPU, one CPU core is used per simulation.
In contrast to the good performance of normal MD simulations, TI simulations with Amber 20 can currently only utilize the GPU by approx. 40-50 % for known algorithmic reasons,[4] though still offering a great performance increase compared to CPU-only implementations and other MD codes. A simple workaround for this performance problem was developed together with the RRZE NHR/HPC group.[5] It allows to make better use of the GPU (up to 99 %) and thus helps to overall increase simulation speed by a factor of approx. 2.8 by using the Cuda Multi-Process Service (MPS)[6] and running several pmemd.cuda jobs (e.g. 5 or 6 λ-windows in our case) at the same time in parallel on one GPU (plus one CPU core per pmemd.cuda process). With this approach, the expensive hardware can be used more efficiently, and longer simulation times and thus better sampling and convergence can be achieved.
Results
Our simulations of the intrahelical TDG-DNA complexes show that discrimination between the four damaged DNA bases cannot take place before the lesioned base is flipped out into the enzyme’s active site and that imino tautomers of the bases do not play a role in substrate recognition at this stage.[7]
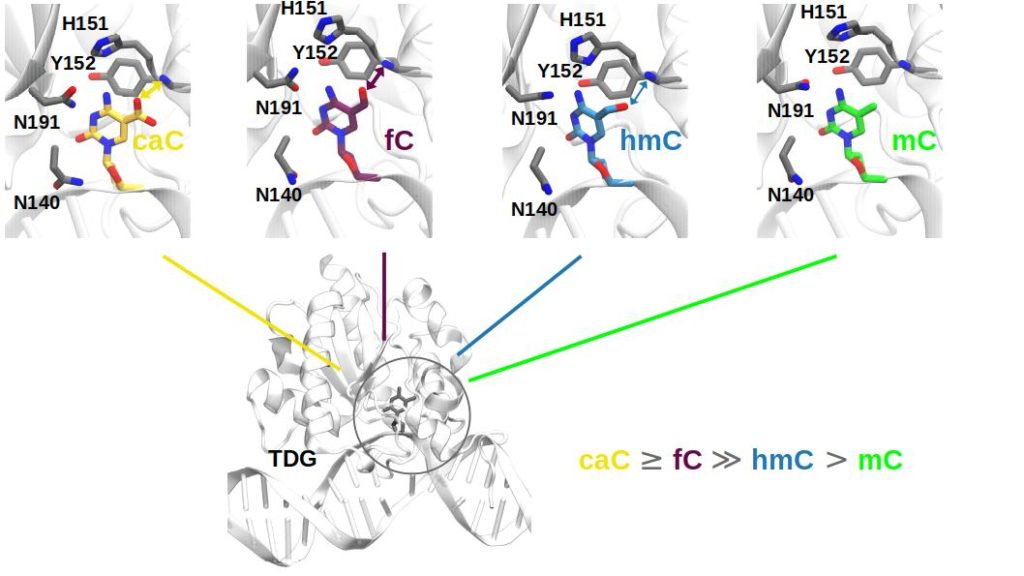
When the damaged base has been flipped out into the active site, there are, however, pronounced differences between the complexes with caC, fC, hmC and mC, both in terms of binding pocket interactions and binding free energies. The cognate bases caC and fC bind much better to TDG than the non-cognate bases hmC and mC and important interactions of the base with active side residues of the protein are only observed for caC and fC. Additionally, a protonated H151 (see Figure 1) is likely for the caC-TDG complex. This additional proton is essential for the subsequent chemical step for caC. Water molecules as potential nucleophiles are also readily available for the subsequent chemical step. Overall, our simulations show that substrate specificity for fC and caC is at least partially achieved by the favorable binding to TDG in an extrahelical complex.[8]
Outreach
F. Beierlein, S. Volkenandt, P. Imhof, Oxidation Enhances Binding of Extrahelical 5-Methyl-Cytosines by Thymine DNA Glycosylase, J. Phys. Chem. B 2022, 126, 1188-1201.
S. Volkenandt, F. Beierlein, P. Imhof, Interaction of Thymine DNA Glycosylase with Oxidised 5-Methyl-cytosines in Their Amino- and Imino-Forms, Molecules 2021, 26, 5728.
Researcher’s Bio and Affiliation
Frank Beierlein studied chemistry and obtained his Ph.D. in Tim Clark’s group in Erlangen in 2005, where he used classical MD simulations and QM/MM calculations to investigate biomolecular systems and their spectroscopic properties. From 2006 to 2008, he worked in the group of Jonathan Essex at the University of Southampton on perturbation free energy methods of proteins and their complexes, also in combination with QM/MM. In 2008, he returned to Erlangen, where he continued his research on biomolecular systems and also material systems, in addition to teaching activities. In the past years, Frank’s interests shifted increasingly to simulations of nucleic acids and their complexes with proteins. He joined Petra Imhof’s group at the CCC in Erlangen in 2020, and NHR@FAU as a liaison scientist in 2021.
References
[1] Case, D. A.; Belfon, K.; Ben-Shalom, I. Y.; Brozell, S. R.; Cerutti, D. S.; Cheatham III, T. E.; V.W.D. Cruzeiro; Darden, T. A.; Duke, R. E.; Giambasu, G.; Gilson, M. K.; Gohlke, H.; Goetz, A. W.; Harris, R.; Izadi, S.; Kasavajhala, K.; Kovalenko, A.; Krasny, R.; Kurtzman, T.; Lee, T. S.; LeGrand, S.; Li, P.; Lin, C.; Liu, J.; Luchko, T.; Luo, R.; Man, V.; Merz, K. M.; Miao, Y.; Mikhailovskii, O.; Monard, G.; Nguyen, H.; Onufriev, A.; Pan, F.; Pantano, S.; Qi, R.; Roe, D. R.; Roitberg, A.; Sagui, C.; Schott-Verdugo, S.; Shen, J.; Simmerling, C. L.; Skrynnikov, N.; Smith, J.; Swails, J.; Walker, R. C.; Wang, J.; Wilson, L.; Wolf, R. M.; Wu, X.; York, D. M.; Kollman, P. A. Amber 2020, University of California: San Francisco, 2020.
[2] Amber Web Page. http://ambermd.org/.
[3] Tutorial 7.3 Thermodynamic Integration Using Soft Core Potentials/Side–Chain Mini Tutorial, Amber Web Site. http://ambermd.org/tutorials/advanced/tutorial9/index.html#sidechain_mini.
[4] Lee, T.-S.; Allen, B. K.; Giese, T. J.; Guo, Z.; Li, P.; Lin, C.; McGee, T. D.; Pearlman, D. A.; Radak, B. K.; Tao, Y.; Tsai, H.-C.; Xu, H.; Sherman, W.; York, D. M. Alchemical Binding Free Energy Calculations in Amber20: Advances and Best Practices for Drug Discovery. J. Chem. Inf. Model. 2020, 60 (11), 5595-5623. doi:10.1021/acs.jcim.0c00613.
[5] Zeiser, T. Personal Communication.
[6] https://docs.nvidia.com/deploy/pdf/CUDA_Multi_Process_Service_Overview.pdf.
[7] Volkenandt, S.; Beierlein, F.; Imhof, P. Interaction of Thymine DNA Glycosylase with Oxidised 5-Methyl-Cytosines in Their Amino- and Imino-Forms. Molecules 2021, 26 (19), 5728. doi:10.3390/molecules26195728.
[8] Beierlein, F.; Volkenandt, S.; Imhof, P. Oxidation Enhances Binding of Extrahelical 5-Methyl-Cytosines by Thymine DNA Glycosylase. J. Phys. Chem. B 2022, 126 (6), 1188-1201. doi:10.1021/acs.jpcb.1c09896.