HPC User Report from S. Ninova (Theoretical Solid-State Physics)
Porphyrin-Oxide Interaction at the Surface
Porphyrins are attractive molecules for solar-to-energy and catalytical applications as part of organic-inorganic devices. Understanding thus how the two phases interacts enables us to fine-tune and improve their performance.
Motivation and problem definition
Recently, it was observed that a monolayer of Co(II) tetraphenylporphyrin (CoTPP), a member of the family of metalloporhyrins, forms stronger and weaker bonds upon interaction with the MgO/Ag(100) surface as compared to the multilayer [Franke et al., Phys. Chem. Chem. Phys., 2017, 19, 11549-11553]. We looked into how the different surface adsorption sites affect the molecular electronic structure from a theoretical point of view.
Methods and codes
We performed calculations within the framework of density-functional theory (DFT) for the geometry relaxation and electronic structure. Additional many-body perturbation theory calculations within the GW approach were carried out, in order to capture physical effects missing in the standard DFT. All simulations were done with Vienna Ab-initio Simulation Package (VASP).
Results
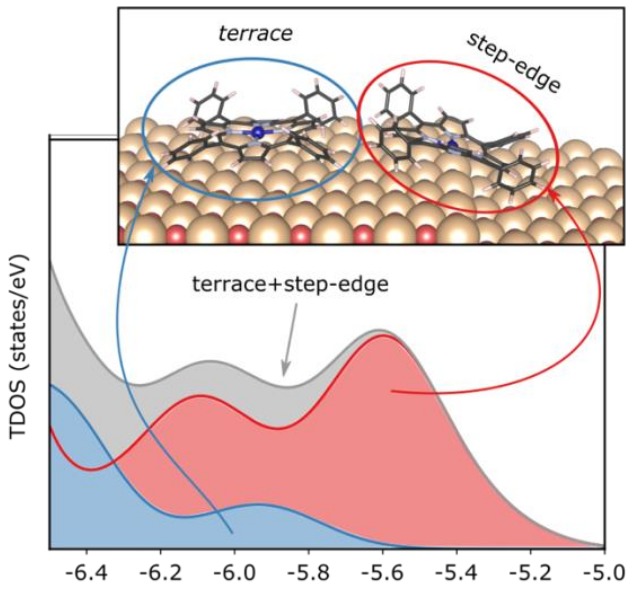
The interaction between CoTPP and MgO(100) surface strongly depends on the adsorption site. On such surfaces, flat terraces and steps coexist. The porphyrin molecules bind more strongly to Figure 1: Differences in the total density of states for the CoTPP the these latter low-coordinated sites as on flat and stepped MgO(100). opposed to those on the flat terrace. This enhanced interaction is accompanied by a strong Co-O bond in the valence band (see Figure). Upon the application of a correction scheme for the short-comings of DFT calculations, we simulated photoemission spectra and were able to assign the experimentally observed features to the adsorption sites.
Outreach
The work is part of the FAU funCOS project (FOR 1878) and one manuscript is in preparation. The results were presented at the Joint Swiss and Austrian Physical Societies meeting in Zürich (2019) and at the 2 nd funCOS International Workshop in Erlangen (2019).
Researcher’s Bio and Affiliation
Silviya Ninova obtained a bachelor’s degree in chemistry and a master’s degree in computational chemistry from Sofia University (Bulgaria). She obtained a doctoral degree from the University of Florence (Italy) and went on to do post-doctoral studies at the University of Bern (Switzerland). Currently, she works as a post-doctoral researcher at the Paris-Lodron University of Salzburg with Prof. Michel Bockstedte (now JKU Linz (Austria) and also Chair for Theoretical Solid-State Physics, FAU).