Molecular dynamics (MD) simulations can be used to calculate various thermophysical properties by studying the statistical motion of molecules and atoms. MD simulations have been used in recent years at the Institute of Advanced Optical Technologies-Thermophysical Properties (AOT-TP), which is headed by Prof. Andreas Paul Fröba and located at the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), for thermophysical property research. Here, simulations are combined with experimental techniques, such as dynamic light scattering (DLS), for the determination of several thermophysical properties of working fluids in chemical and energy engineering. MD simulations have been applied for the development of a fundamental understanding of how the molecular characteristics or fluid structure influence the properties of pure fluids and of binary and ternary mixtures.
Motivation and problem definition
For an efficient design and for the optimization of processes related to chemical and energy engineering, the thermophysical properties of the working fluids, which may contain multiple species including dissolved gases, have to be known. One challenge related to the determination of thermophysical properties might be the relatively large temperatures T and pressures p at which, for example, chemical reactions take place. But also, the handling of flammable and toxic substances might hinder an experimental investigation. An alternative to experiments, where due to the aforementioned challenges substantial resources and efforts are required, are MD simulations. The accuracy of MD simulations to predict various thermophysical properties is, however, highly dependent on the underlying force field (FF), which describes how atoms within the same molecule or between two molecules can interact with each other. It is, therefore, imperative that the capabilities of commonly used FFs to predict the thermophysical properties of interest are tested. Additionally, a check of the transferability to further substances, which were not included in the FF development is necessary. Here, results from the experiments at the same thermodynamic states are required to serve as a reference. Especially at elevated T and p and for kinetic and transport properties, e.g., viscosity and various diffusivities, experimental data are still scarce in the literature, which makes the validation and development of FFs challenging.
Next to the determination of thermophysical properties, MD simulations provide an insight into the fluid structure on a molecular level. This allows to investigate the influence of intermolecular interactions, e.g., in the form of hydrogen bonding, on the thermophysical properties and to establish structure-property relationships, which are often the basis for the development of prediction models for thermophysical properties.
Methods and codes
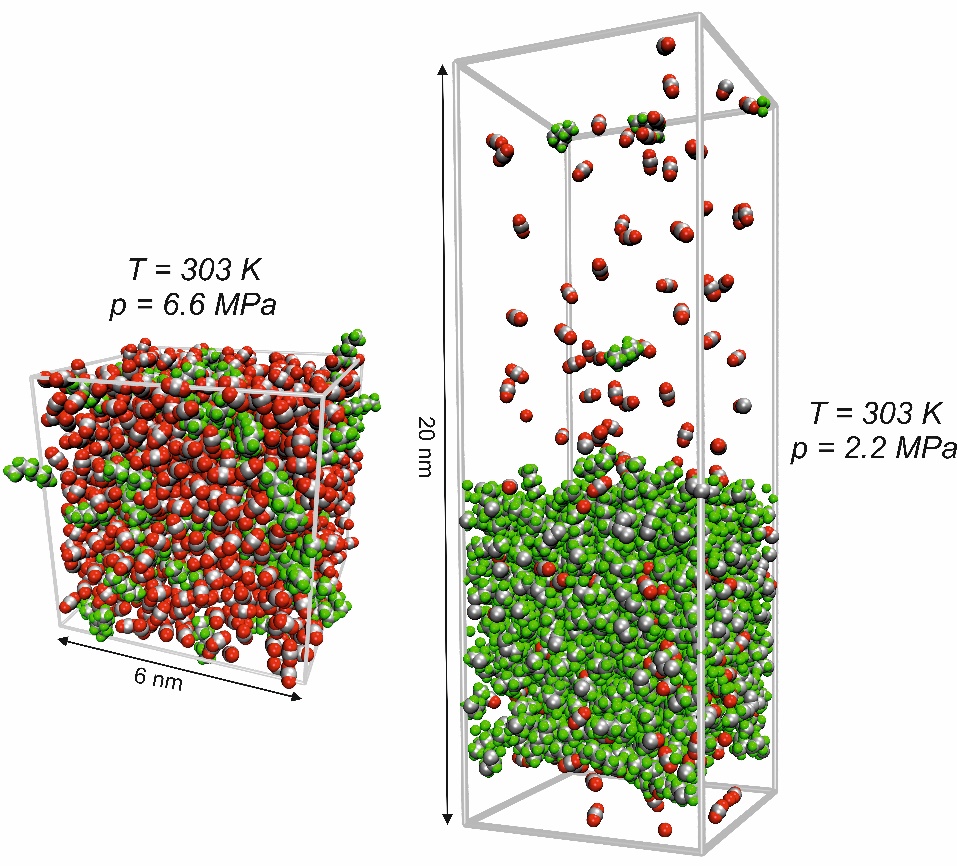
The GROMACS software package [1] is used for performing equilibrium molecular dynamics (EMD) simulations in macroscopic thermodynamic equilibrium in the vapor or liquid phase as well as at vapor-liquid equilibrium with typically 30,000 atoms, c.f. Figure 1. To calculate, for example, density, interfacial tension, or radial-distribution functions, corresponding post-processing tools included in the GROMACS package are used. For the computation of kinetic and transport properties such as dynamic viscosity, self-diffusivity, or Maxwell-Stefan diffusivity from the statistical motion of the molecules, post-processing tools were developed in-group. Here, mainly FORTRAN codes are used to handle the large amounts of data necessary for calculating properties from statistical fluctuations.
Results
Within the DFG-funded research projects FR 1709/11-1, FR 1709/11-2, and FR 1709/12-1, the mutual diffusion coefficient D11 could be determined from EMD simulations in the superheated vapor phase as well as in the vapor and liquid phase close to saturation conditions [P1-P3, P6-P8, P11, P13, P17]. The investigated systems consist of linear and branched alkanes and alcohols with a large variation of the carbon-chain length, ionic liquids (ILs), and various gases including refrigerants. Within the aforementioned projects, binary mixtures of gases or liquids as well as binary mixtures consisting of a liquid and a dissolved gas were investigated. The self-developed codes were used to determine besides the Maxwell-Stefan diffusivity (Đ12) also the thermodynamic factor (Γ11). Both give access to D11 according to D11 = Đ12 ∙ Γ11 in a purely predictive manner without the use of a system-specific binary interaction parameter, c.f. Figure 2. The EMD simulations could be validated with the help of results from DLS and agreement was found typically within 15%. With the help of a binary mixture consisting of an IL and dissolved carbon dioxide, for the first time, the fully predictive determination of D11 from EMD simulations could be demonstrated for an electrolyte system, where, due to the dissociation of the IL into anions and cations, the determination of Đ12 and Γ11 is more complex [P13]. Here, the ability of the simulations to analyze the fluid structure on a molecular level, which was further validated by Raman spectroscopy, was indispensable in describing how the formation of ion pairs influences D11 [P13]. Based on the results from EMD simulations and DLS for D11 in mixtures consisting of a liquid with a dissolved gas at infinite dilution, a simple prediction model could be developed [P2, P6]. The model requires only pure component properties of the solvent and solute and can predict D11 with an average absolute relative deviation of about 12%.
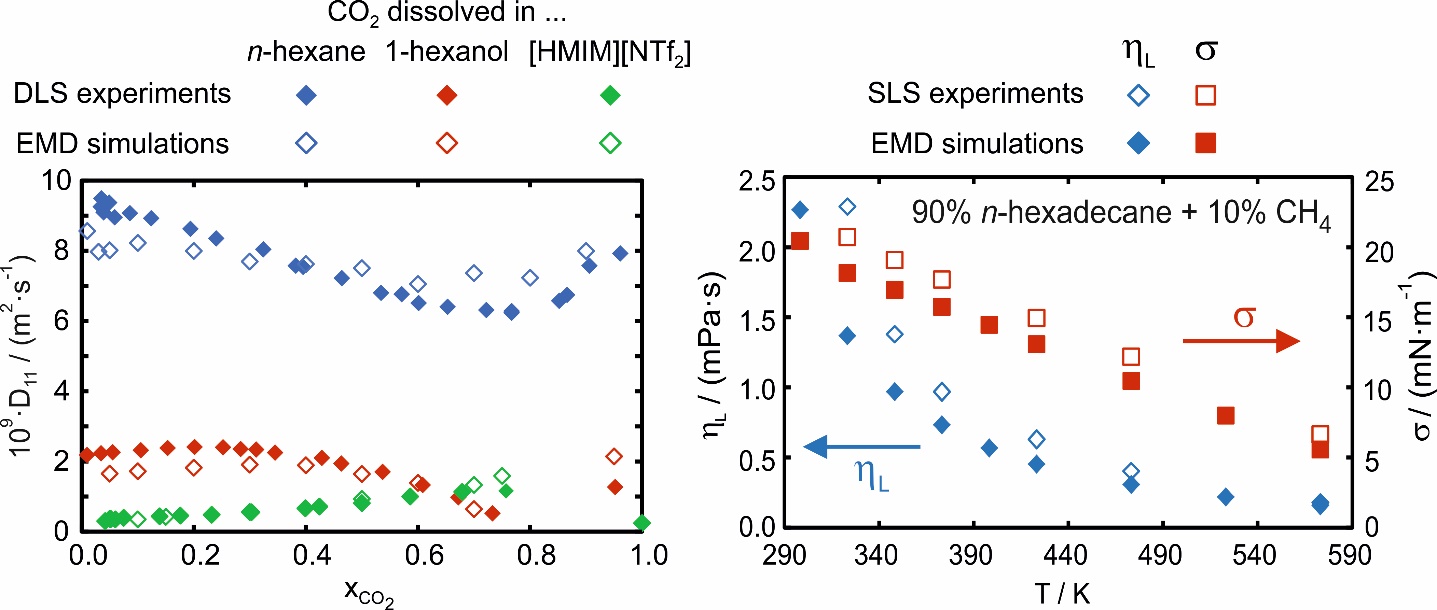
In the further DFG-funded project FR 1709/15-1, the focus was on the determination of the dynamic viscosity η and interfacial tension σ [P9-P10, P12, P14-P16, P18-P19]. For long linear and branched alkanes and alcohols as well as LOHCs, commonly applied FFs from the literature have shown, especially at large T, large deviations up to 100% when predicting η and σ. Therefore, a T-dependent correction term for the L-OPLS FF [2-3] was developed and included in the description of dispersive and repulsive interactions between atoms [P9]. With the modified L-OPLS FF, the T-dependent drift of the simulated properties with respect to the experimental ones could be resolved and the agreement could be improved [P9, P10]. Using the modified L-OPLS FF, the influence of dissolved gases on the thermophysical properties of alkanes, alcohols, and LOHCs could be investigated, c.f. Figure 2. Also here, the results from DSL applied to the vapor-liquid interface, often referred to as surface light scattering (SLS), were used to validate the simulations.
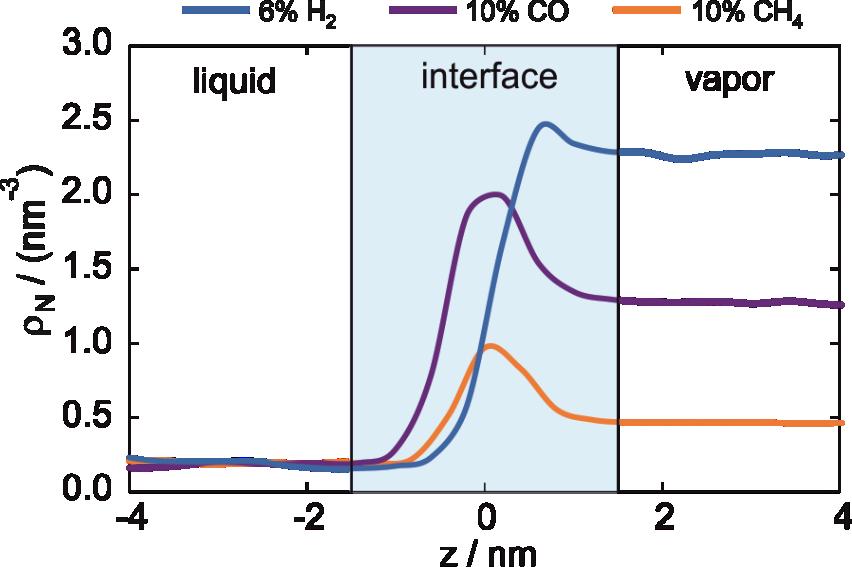
With the help of the pure component properties from SLS and EMD simulations, the influence of the dissolved gas could be determined. Especially for σ, the ability of EMD simulations to analyze the fluid structure on a molecular level was invaluable to fundamentally understand how the molecular characteristics influence the property [P14, P16, P18-P19]. With the help of spatially resolved partial number densities ρ, the enrichment of the gas molecules could be utilized to understand why different gas molecules influence σ differently at comparable T, p, and composition, c.f. Figure 3.
Outreach
The results from the various DFG-funded projects (FR 1709/11-1, FR 1709/11-2, FR 1709/12-1, and FR 1709/15-1) have been published in multiple peer-reviewed journals. Additionally, the results have been presented at national and international conferences, such as the “Thermodynamik-Kolloquium” in the years 2017-2019 and 2021-2022, the “European Conference on Thermophysical Properties” in 2017, the “Symposium on Thermophysical Properties” in 2018 and 2021 in Boulder, USA, and the “Asian Thermophysical Properties Conference” in 2022.
Publications acknowledging the computational resources provided by the RRZE in peer-reviewed journals:
[P1] Klein, T., Wu, W., Rausch, M. H., Giraudet, C., Koller, T. M., Fröba, A. P. Influence of Liquid Structure on Fickian Diffusion in Binary Mixtures of n-Hexane and Carbon Dioxide Probed by Dynamic Light Scattering, Raman Spectroscopy, and Molecular Dynamics Simulations. J. Phys. Chem. B 122 (2018), p. 7122-7133.
[P2] Giraudet C., Klein T., Zhao G., Rausch M. H., Koller T. M., Fröba A. P. Thermal, Mutual, and Self-Diffusivities of Binary Liquid Mixtures Consisting of Gases Dissolved in n-Alkanes at Infinite Dilution. J. Phys. Chem. B 122 (2018), p. 3163-3175.
[P3] Higgoda U., Hewa Kankanamge C. J. K., Hellmann R., Koller T. M., Fröba A. P. Fick Diffusion Coefficients of Binary Fluid Mixtures Consisting of Methane, Carbon Dioxide, and Propane via Molecular Dynamics Simulations Based on Simplified Pair-Specific ab Initio-Derived Force Fields. Fluid Phase Equilib. 502 (2019), Article No.: 112257.
[P4] Higgoda U., Hellmann R., Koller T. M., Fröba A. P. Self-Diffusion Coefficient and Viscosity of Methane and Carbon Dioxide via Molecular Dynamics Simulations Based on New ab Initio-Derived Force Fields. Fluid Phase Equilib. 481 (2019), p. 15-27.
[P5] Higgoda U., Hellmann R., Koller T. M., Fröba A. P.: Enhancement of the Predictive Power of Molecular Dynamics Simulations for the Determination of Self-Diffusion Coefficient and Viscosity Demonstrated for Propane. Fluid Phase Equilib. 496 (2019), p. 69-79.
[P6] Wu, W., Klein, T., Kerscher, M., Rausch, M. H., Koller, T. M., Giraudet, C., Fröba, A. P. Diffusivities in 1-Alcohols Containing Dissolved H2, He, N2, CO, or CO2 Close to Infinite Dilution. J. Phys. Chem. B 123 (2019), p. 8777–8790.
[P7] Wu W., Klein T., Kerscher M., Rausch M. H., Koller T. M., Giraudet C., Fröba A. P. Mutual and Thermal Diffusivities as Well as Fluid-Phase Equilibria of Mixtures of 1-Hexanol and Carbon Dioxide. J. Phys. Chem. B 124 (2020), p. 2482-2494.
[P8] Klein T., Piszko M., Lang M., Mehler J., Schulz P., Rausch M. H., Giraudet C., Koller T. M., Fröba A. P. Diffusivities in Binary Mixtures of [AMIM][NTf2] Ionic Liquids with the Dissolved Gases H2, He, N2, CO, CO2, or Kr Close to Infinite Dilution. J. Chem. Eng. Data 65 (2020), p. 4116–4129.
[P9] Klein T., Lenahan F., Kerscher M., Rausch M. H., Economou I. G., Koller T. M., Fröba A. P. Characterization of Long Linear and Branched Alkanes and Alcohols for Temperatures up to 573.15 K by Surface Light Scattering and Molecular Dynamics Simulations. J. Phys. Chem. B 124 (2020), p. 4146-4163
[P10] Kerscher, M., Klein, T., Schulz, P., Veroutis, E., Dürr, S., Preuster, P., Koller, T. M., Rausch, M. H., Economou, I. G., Wasserscheid, P., Fröba, A. P. Thermophysical Properties of Diphenylmethane and Dicyclohexylmethane as a Reference Liquid Organic Hydrogen Carrier System from Experiments and Molecular Simulations. Int. J. Hydrog. Energy, 45 (2020), p. 28903-28919.
[P11] Piszko M., Lenahan F., Hahn S., Rausch M. H., Koller T. M., Klein T., Fröba A. P. Diffusivities in Binary Mixtures of I-Hexane or 1-Hexanol with Dissolved CH4, Ne, Kr, R143a, SF6, or R236fa Close to Infinite Dilution. J. Chem. Eng. Data 66 (2021), p. 2218-2232.
[P12] Lenahan F., Zikeli M., Rausch M. H., Klein T., Fröba A. P. Viscosity, Interfacial Tension, and Density of Binary-Liquid Mixtures of n-Hexadecane with n-Octacosane, 2,2,4,4,6,8,8-Heptamethylnonane, or 1-Hexadecanol at Temperatures between 298.15 and 573.15 K by Surface Light Scattering and Equilibrium Molecular Dynamics Simulations. J. Chem. Eng. Data 66 (2021), p. 2264-2280.
[P13] Klein, T., Piszko, M., Kankanamge, C. J., Kasapis, G., Fröba, A. P. Fick Diffusion Coefficient in Binary Mixtures of [HMIM][NTf2] and Carbon Dioxide by Dynamic Light Scattering and Molecular Dynamics Simulations. J. Phys. Chem. B 125 (2021), p. 5100-5113
[P14] Klein T., Lenahan F., Kerscher M., Jander J. H., Rausch M. H., Koller T. M., Fröba A. P. Viscosity and Interfacial Tension of Binary Mixtures of n-Hexadecane with Dissolved Gases Using Surface Light Scattering and Equilibrium Molecular Dynamics Simulations. J. Chem. Eng. Data 66 (2021), p. 3205-3218.
[P15] Schmidt P., Kerscher M., Klein T., Jander J. H., Bioucas F., Rüde T., Li S., Stadelmaier M., Hanyon S., Fathalla R., Bösmann A., Preuster P., Wasserscheid P., Koller T. M., Rausch M. H., Fröba A. P. Effect of the Degree of Hydrogenation on the Viscosity, Surface Tension, and Density of the Liquid Organic Hydrogen Carrier System Based on Diphenylmethane. Int. J. Hydrog. Energy 47 (2022), p. 6111-6130.
[P16] Lenahan F., Zhai Z., Hewa Kankanamge C. J. K., Klein T., Fröba A. P. Viscosity and Interfacial Tension of Ternary Mixtures Consisting of Linear Alkanes, Alcohols, and/or Dissolved Gases Using Surface Light Scattering and Equilibrium Molecular Dynamics Simulations. Int. J. Thermophys. 43 (2022), Article No.: ARTN 116.
[P17] Lenahan, F., Piszko, M., Klein, T., Fröba, A. P. Diffusivities in Binary Mixtures of n-Decane, n-Hexadecane, n-Octacosane, 2-Methylpentane, 2,2-Dimethylbutane, Cyclohexane, Benzene, Ethanol, 1-Decanol, Ethyl Butanoate, or n-Hexanoic Acid with Dissolved He or Kr Close to Infinite Dilution. J. Chem. Eng. Data, 67 (2022), p. 622-635.
[P18] Klein T., Lenahan F., Zhai Z., Kerscher M., Jander J. H., Koller T. M., Rausch M. H., Fröba A. P. Viscosity and Interfacial Tension of Binary Mixtures Consisting of Linear, Branched, Cyclic, or Oxygenated Hydrocarbons with Dissolved Gases Using Surface Light Scattering and Equilibrium Molecular Dynamics Simulations. Int. J. Thermophys. 43 (2022), Article No.: 88.
[P19] Kankanamge, C. J. Lenahan, F., Klein, T., Fröba, A. P. Viscosity and Interfacial Tension of Binary Mixtures Consisting of an n-Alkane, Branched Alkane, Primary Alcohol, or Branched Alcohol and a Dissolved Gas Using Equilibrium Molecular Dynamics Simulations. Int. J. Thermophys. 43 (2022), Article No.: ARTN 112.
References
[1] Abraham, M. J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J. C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations Through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1-2, 19-25.
[2] Siu, S. W. I.; Pluhackova, K.; Böckmann, R. A. Optimization of the OPLS-AA Force Field for Long Hydrocarbons. J. Chem. Theory Comput. 2012, 8, 1459-1470.
[3] Pluhackova, K.; Morhenn, H.; Lautner, L.; Lohstroh, W.; Nemkovski, K. S.; Unruh, T.; Böckmann, R. A. Extension of the LOPLS-AA Force Field for Alcohols, Esters, and Monoolein Bilayers and its Validation by Neutron Scattering Experiments. J. Phys. Chem. B 2015, 119, 15287-15299.
Researcher’s Bio and Affiliation
Tobias Klein obtained his Master of Science (M.Sc.) degree in Energy Technology at the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU). Since April 2016, he is working a doctoral candidate in the Institute of Advanced Optical Technologies – Thermophysical Properties (AOT-TP) under supervision of Prof. Dr.-Ing. habil. Andreas P. Fröba.